Profil génétique du chat – Cardiomyopathie hypertrophique (CMH)

Rédaction par Dr David W. Silversides, DVM
Traduction par Dr Antoine Cournoyer, DMV
Tout comme chez l’humain, la cardiomyopathie hypertrophique (CMH) est l’une des maladies du cœur les plus courantes observées cliniquement chez les chats. Chez ces deux espèces, les symptômes comprennent un épaississement pathologique de la paroi du muscle du coeur (hypertrophie), une arythmie, des embolies obstructives, un essoufflement, une intolérance à l’exercice, une insuffisance cardiaque et le potentiel d’une mort cardiaque soudaine. Des études chez les humains ont révélé que la CMH est causée par un problème fonctionnel et structurel au niveau des fibres musculaires. Cette maladie se présente comme une maladie génétique impliquant une transmission autosomique dominante. En effet, plus de 1 400 mutations dans au moins onze gènes différents ont été détectées chez l’humain.
Signes cliniques chez le Maine Coon et le Ragdoll

La CMH est présente chez de nombreuses races de chats, mais plus particulièrement chez les Maine Coon et les Ragdoll. L’apparition des signes cliniques varie considérablement et peut survenir entre l’âge d’un et quatre ans, voire plus tard dans la vie du chat. Un souffle cardiaque ausculté au stéthoscope et une hypertrophie du cœur évidente à l’examen échographique figurent parmi les premiers signes cliniques de la maladie. Les symptômes peuvent être légers, modérés ou sévères et ils peuvent se stabiliser ou progresser. De façon générale, les symptômes de la CMH apparaissent plus tôt et sont plus sévères chez les mâles que chez les femelles.
La mutation génétique du CMH
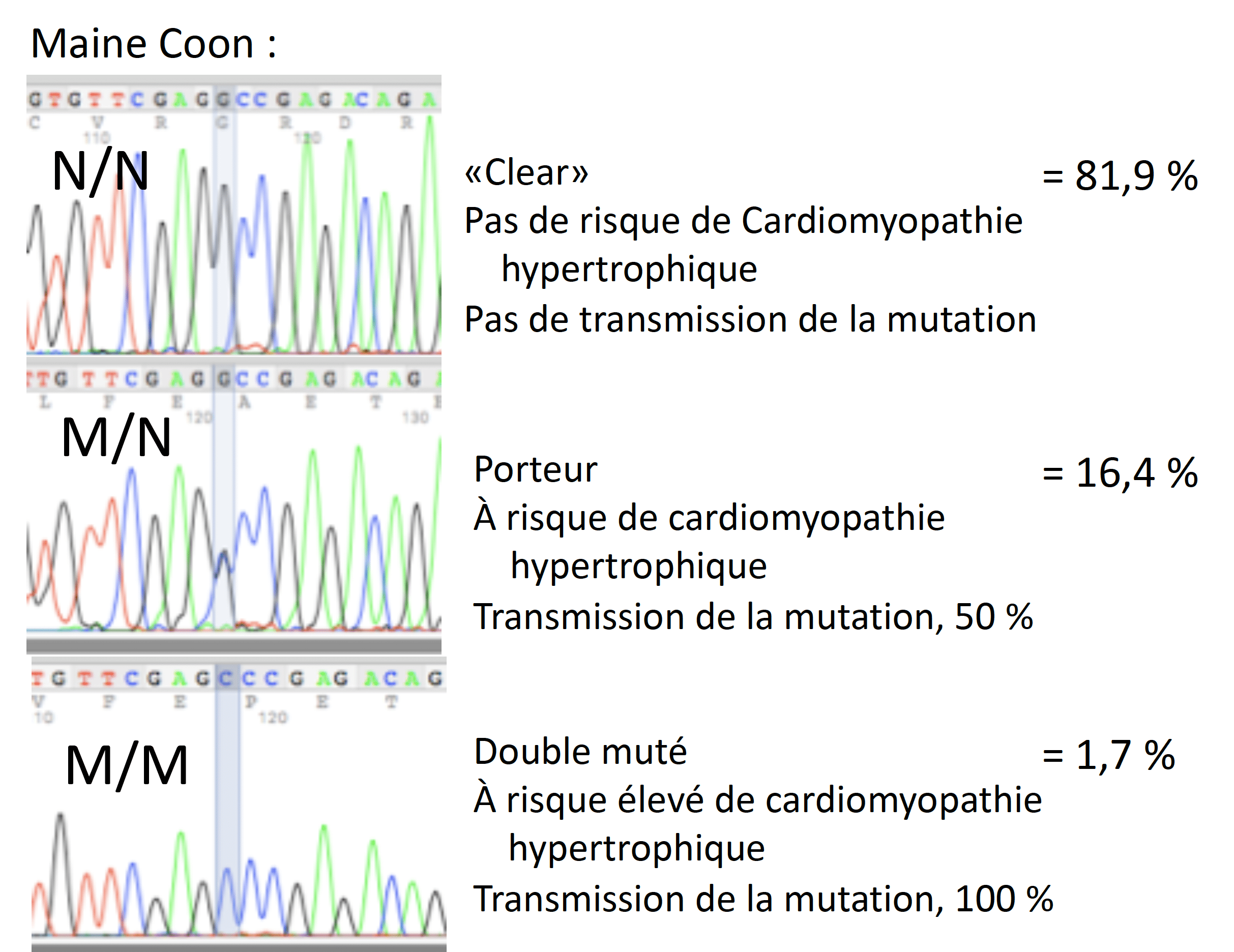
Les connaissances de la CMH chez les humains ont été utiles pour identifier deux mutations distinctes du gène MYBPC3 chez les chats. Ce gène code pour une protéine régulatrice du myocarde qui contribue à contrôler la vitesse et la force de contraction. En fait, les mutations au niveau du gène MYBPC3 sont un facteur important du développement de la CMH chez les humains. Chez les Maine Coon, la mutation a lieu lorsque la lettre génétique (base) G est remplacée par un C (Meurs 2005). Les premières études ont révélé que 31% des Maine Coon étaient porteurs de cette mutation (M/N hétérozygote), tandis que 3% présentaient deux mutations (M/M homozygote dominant) (Fries 2008). À ce jour, parmi les échantillons soumis au laboratoire Labgenvet provenant des chats Maine Coon, 81,9% d’entre eux étaient normaux (N/N), 16,4% étaient porteurs (M/N) et 1,7% étaient M/M (double muté).
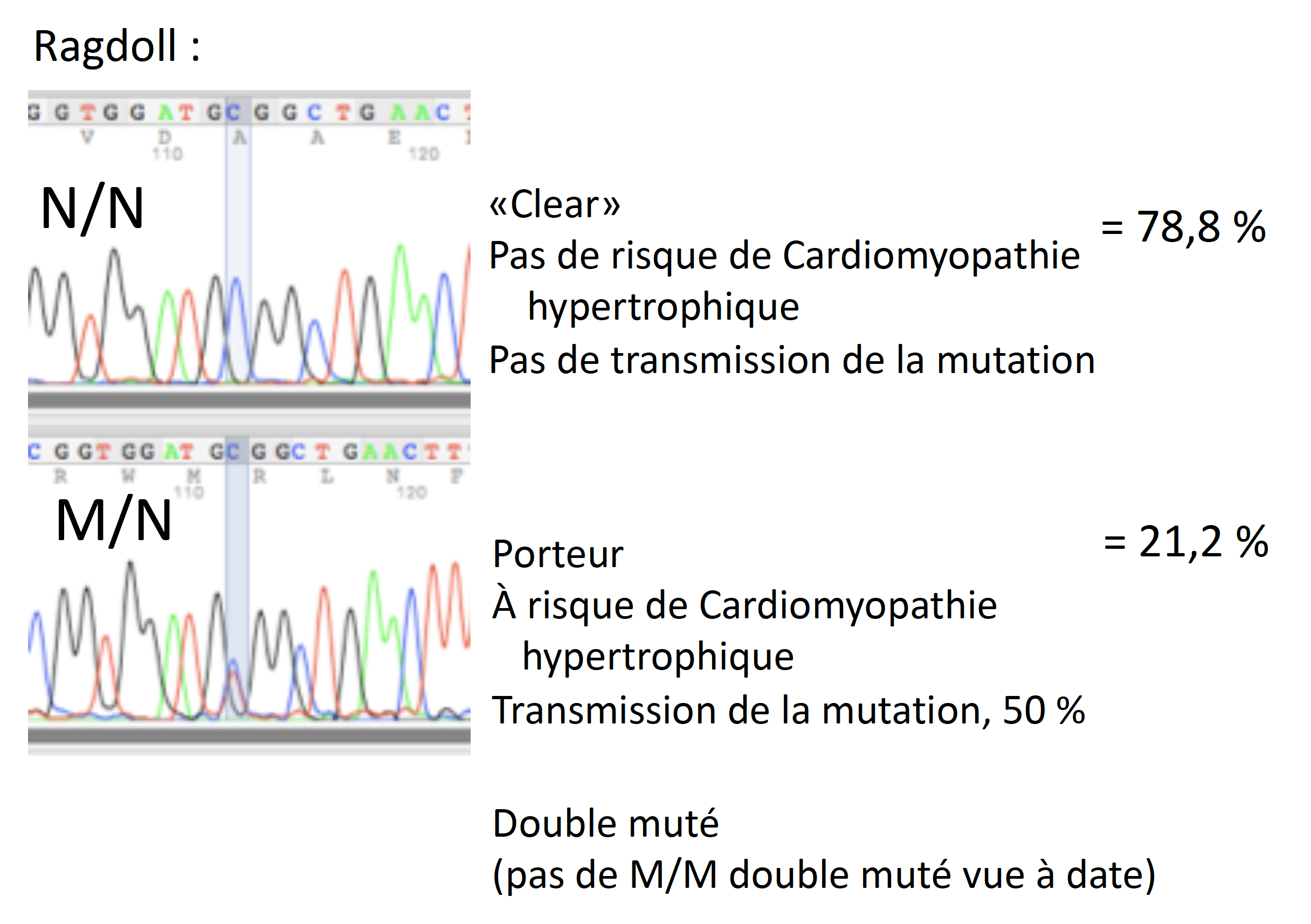
La mutation se situe à un autre endroit sur le gène chez les Ragdoll comparativement au Maine Coon. La lettre C est remplacée par un T (Meurs 2007). La fréquence de cette mutation est similaire à celle chez le Maine Coon. (Borgeat 2014). À ce jour, parmi les échantillons soumis au laboratoire Labgenvet provenant des chats Ragdolls, 78,8% d’entre eux étaient normaux (N/N) et 21,2% étaient porteurs (M/N). Aucun animal présentant deux mutations (M/M) n’a été recensé.
La génétique compliquée
Autant chez le Ragdoll que chez le Maine Coon, un chat présentant deux mutations (M/M) a plus de chance de développer la CMH (taux de pénétration élevé à plus de 50%). Lorsque l’animal a une mutation sur une seule copie de son gène (M/N), les chances de développer la CMH sont nettement plus basses (faible taux de pénétration autour de 8%). D’ailleurs, certains chats de races Maine Coon, Ragdoll ou autres développent la CMH sans être porteurs des mutations connues au niveau du gène MYBPC3. Autrement dit, cela indique que, même si la CMH est une maladie génétique dominante, tout comme chez l’humain, l’implication génétique de la maladie chez le chat n’est pas si simple. Il existe sans aucun doute d’autres mutations causant la CMH qui n’ont pas encore été découvertes, ainsi que d’autres gènes modificateurs qui affectent la fonction des gènes connus.
Identifier les animaux porteurs

Les raisons pour lesquelles les mutations connues qui causent la CMH sont relativement fréquentes chez les Maine Coon et les Ragdoll ne sont pas bien comprises. Le fait que les symptômes se présentent souvent chez les animaux utilisés pour la reproduction illustre qu’il y a eu peu de sélection naturelle pour éliminer la mutation. De plus, étant donné le faible taux de pénétration de la maladie chez les animaux porteurs (M/N), il est difficile de reconnaître cliniquement les porteurs. Un test d’ADN pour l’identification des mutations responsables de la CMH chez les Maine Coon et les Ragdoll permet de mieux reconnaître les animaux porteurs et aident à réduire la fréquence des mutations au fil du temps. Comme les mutations sont relativement fréquentes et que le taux de pénétration est faible chez les chats porteurs (M/N), quelques pratiques de reproduction sont recommandées. La reproduction de chats présentant deux mutations (M/M) est donc déconseillée parce que la mutation ainsi que le risque de développer la maladie sont ainsi transmis. L’accouplement d’un chat porteur (M/N) avec un chat clear (N/N) est acceptable pendant quelques générations, pour ne pas réduire le bassin génétique. Éventuellement, à l’aide des tests d’ADN, les animaux porteurs pourront être éliminés de la reproduction.
References:
- Meurs KM, Sanchez X, David RM et al. (2005) A cardiac myosin binding protein C mutation in the Maine coon cat with familial hypertrophic cardiomyopathy. Hum. Mol. Genet. 14:3587-93. [pubmed/16236761]
- Meurs KM, Norgard MM et al. (2007) A substitution mutation in the myosin binding protein C gene in ragdoll hypertrophic cardiomyopathy. Genomics 90(2) :261-64. [pubmed/17521870]
- Fries R, Heaney AM, Meurs KM. (2008) Prevalence of the myosin-binding protein C mutation in Maine Coon cats. J Vet Intern Med 22:893-896. [pubmed/18498321]
- Borgeat K, Casamian-Sorrosal D, Helps C et al. (2014) Association of the myosin binding protein C3 mutation (MYBPC3 R820W) with cardiac death in a survey of 236 Ragdoll cats. J Vet Cardiol 16(2) :73-80. [pubmed/24906243]
- Kittleson MD, Meurs KM, Harris SP (2015) The genetic basis of hypertrophic cardiomyopathy in cats and humans. J Vet Cardio 17:S53-S73. [pubmed/26776594]
